Régions codantes
Ce projet explore la recherche de sous-mots, appliquée à l’analyse de séquences génomiques. Il s’inscrit dans le cadre d’un travail pratique réalisé lors du module d’informatique de mon master Mathématiques et Applications à l’EAD de l’Université d’Aix-Marseille. Le travail demandé s’inspire directement de l’article À la recherche de régions codantes, rédigé par François Rechenmann et publié sur la revue en ligne Interstices. Cet article éclaire les méthodes algorithmiques employées pour identifier des motifs significatifs dans les séquences d’ADN, et sa lecture est vivement recommandée pour approfondir le sujet.
Choix d’objets de manipulation dans les fonctions:
On essaye d’utiliser des tableau de bitarray et faire des calculs et opération dessus au lieu de tableau d’entiers.
Comme chaque entier ou caractère du type char occupe 28 octets; utiliser une alternative bitarray(), où chaque nucléotide ne prend que 2 bits au lieu de 28 octets me semblait intéressant à explorer. Problèmes avec ce choix : pour utiliser un dictionnaire avec des clés de type bitarray (ou précisément frozenbitarray), ça s’est avéré compliqué. Un autre inconvénient est dans le fait que la construction d’un bitarray() demande des ressources à chaque fois donc pour bien cerner l’efficacité par rapport à des entiers, il faut peut-etre une comparaison plus approfondie de la complexité et du temps pour chacune de ces méthodes, n’étant pas le focus de ce projet je ne me suis pas lancée dans cette comparaison.
Plus tard j’ai été obligée de revenir aux entiers pour utiliser des methodes numpy, donc utiliser bitarray() n’est peut-etre pas particlulièrement plus intéressant dans ce contexte.
D’un autre côté, danc cette version, j’ai veillé à éviter les boucles et à privilégier la vectorisation, pour tirer parti des bibliothèques optimisées comme numpy et accélérer les calculs.
!pip install bitarray!pip install regexSi on définit le dictionnaire à l’intérieur d’une dataclass Python et que nous définissons les clés en utilisant de conversion à des éléments de bitarray() au moment de la définition, n’est appelé qu’une seule fois pour chaque clé lors de la création de l’objet.
1. Traduction de codons en séquence d’acides aminés.
- Préparation et associations
On définit des dictionnaires qui servent à encoder et décoder les nucléotides et les codons. Par exemple, chaque nucléotide (A, T, C, G) est associé à une représentation binaire sur 2 bits. Un dictionnaire de conversion “inverse” permet de lire la séquence dans l’autre sens (où, par exemple, T devient C et C devient T car 01 se lit 10 au sens inverse). On construit ensuite un arbre de décodage (via la fonction decodetree) qui facilitera revenir en arrière et avoir les bitarrays en séquences de lettres.
-
Construction de l’association canonique Dans la biologie, plusieurs codons (groupes de trois nucléotides) peuvent coder pour le même acide aminé. Pour uniformiser la traduction, le code construit une association canonique ou « mapping canonique » avec la fonction associ_canonique(). Pour chaque codon, il détermine la lettre d’acide aminé correspondante (sauf pour les codons STOP) et choisit la représentation binaire (sur 6 bits) qui a la valeur entière la plus basse. Cette normalisation permet de convertir toutes les variantes synonymes en une seule forme standard.
-
Table de normalisation La fonction table_norm() génère un tableau numpy de taille 64, où chaque indice (de 0 à 63, représente tous les codons possibles en 6 bits) est associé à la valeur entière de son codon canonique. Ce tableau facilite la conversion vectorisée et rapide de codons non normalisés en leurs formes normalisées.
-
Arbre de décodage et encodage des acides aminés Un arbre de décodage est reconstruit à partir de l’association canonique pour décoder les codons en lettres d’acides aminés. Par ailleurs, une association d’encodage convertit chaque acide aminé (sauf STOP) en un bitarray de 5 bits ce qui permet une représentation compacte des protéines.
-
La classe sequenceGenome Cette classe définit une séquence d’ADN sous forme de chaîne de caractères et de bitarray. On y associe des méthodes pour créer un objet à partir d’une chaîne ou directement à partir d’un bitarray.
-
La classe traducteurADN C’est ici que se concentre la traduction proprement dite. Dans son initialisation, le traducteur :
Définit une association complete des codons et des acides aminés. Calcule l’association canonique et la table de normalisation afin de transformer chaque codon en sa forme standard. Construit un arbre de décodage pour retransforme les codons normalisés en lettres. Crée un motif régulier (regex) qui reconnaît soit un codon reconnu (c’est-à-dire 6 bits correspondant à une forme canonique) soit exactement 2 bits à ignorer (pour essayer avec les 6 bits suivant et voir s’il y aurait concordance.)
- La traduction de la séquence
Les méthodes traduire_sequence et traduire_sequence_bits prennent en entrée une séquence ADN (soit dans un objet sequenceGenome, soit directement sous forme de bitarray). Elle effectuent les opérations suivantes :
-
Conversion du bitarray en une chaîne de “0” et “1”.
-
Application du regex pour remplacer chaque codon reconnu par sa forme canonique et éliminer les groupes de bits à ignorer (les 2 bits qui ne font pas partie d’un codon normalisé).
-
Reconstitution d’un bitarray normalisé.
-
Décodage du bitarray normalisé en une séquence d’acides aminés via l’arbre de décodage.
-
Filtrage des acides aminés valides et réencodage en un bitarray protéique, chaque acide aminé étant représenté sur 5 bits.
Une version initiale de cette fonction ou une version alternative (traduire_sequence_bits_v1) réalise la normalisation codon par codon avec une boucle, plutôt que par regex.
import numpy as np
from dataclasses import dataclass
from bitarray import bitarray, frozenbitarray, decodetree
from bitarray.util import int2ba
import regex
#Pour la lecture au sens inverse des séquences
CARTE_INVERSE = {
'A': 'A',
'T': 'C',
'C': 'T',
'G': 'G'
}
# dictionnaire de conversion des nucléotides en 2 bits
CARTE_NUCLEOTIDES = {
'A': frozenbitarray('00'),
'T': frozenbitarray('01'),
'C': frozenbitarray('10'),
'G': frozenbitarray('11')
}
# décodage basé sur le dictionnaire ci dessus
DECODE_TREE = decodetree(CARTE_NUCLEOTIDES)
# carte combinée pour transformation
ASSOCI_COMB = {
CARTE_INVERSE[nuc]: bits
for nuc, bits in CARTE_NUCLEOTIDES.items()
}
# -------------------------------------------------------------------------------------------- Fonctions auxiliaires: ---------------------------------------------------
# établit l'association canonique:
# lettre d'acide aminé -> codon canonique 6 bits type frozenbitarray
def associ_canonique(codons: dict, acides_aminés: dict, carte_nucléotides: dict) -> dict:
association_canonique = {}
for codon in codons:
lettre_aa = acides_aminés[codons[codon]]
if lettre_aa == 'STOP':
continue
bits = frozenbitarray("".join(carte_nucléotides[nt].to01() for nt in codon))
if lettre_aa not in association_canonique or int(bits.to01(), 2) < int(association_canonique[lettre_aa].to01(), 2):
association_canonique[lettre_aa] = bits
return association_canonique
# Construit une tableau numpy de normalisation (taille 64) qui associe chaque codon 6 bits (0-63)
# vers la valeur entière de son codon canonique
def table_norm(codons: dict, acides_aminés: dict, association_canonique: dict, carte_nucléotides: dict) -> np.ndarray:
# tableau d'indices de 0 à 63
indices = np.arange(64, dtype=np.uint8)
# trouver les positions des codons
premier = (indices >> 4) & 3
deuxième = (indices >> 2) & 3
troisième = indices & 3
# association vectorisée des indices vers lettres : 0 -> 'A', 1 -> 'T', 2 -> 'C', 3 -> 'G'
nucleotides = np.array(['A', 'T', 'C', 'G'])
codon_premier = nucleotides[premier]
codon_deuxième = nucleotides[deuxième]
codon_troisième = nucleotides[troisième]
# Concaténer les lettres pour obtenir un tableau (de 64 éléments) de codons par exemple : "ATG")
codons_str = np.char.add(np.char.add(codon_premier, codon_deuxième), codon_troisième)
# Construire le dictionnaire d'association pour les codons connus (sans STOP)
mapping = {
codon: int(association_canonique[acides_aminés[codons[codon]]].to01(), 2)
for codon in codons if acides_aminés[codons[codon]] != 'STOP'
}
# fonction vectorisée : pour chaque codon (s) et son indice (i) retourner mapping[s] si présent, sinon l'indice d'origine i
vec_lookup = np.vectorize(lambda s, i: mapping.get(s, i))
valeurs_norm = vec_lookup(codons_str, indices)
# Retourne un tableau numpy de uint8 contenant les valeurs normalisées
return valeurs_norm.astype(np.uint8)
# Construit l'arbre de décodage à partir du mapping canonique : lettre -> codon canonique
def arbre_decodage(association_canonique: dict):
mapping_decodage = {aa: bits for aa, bits in association_canonique.items()}
return decodetree(mapping_decodage)
# Construit le mapping d'encodage pour les acides aminés : lettre -> bitarray 5 bits (sauf STOP)
def carte_encodage_acides(acides_aminés: dict) -> dict:
return {
aa: int2ba(code, length=5, endian='big')
for code, aa in acides_aminés.items() if aa != 'STOP'
}
# -------------------------------------------------------------- Classe sequenceGenome ------------------------------------------------------#
@dataclass
class sequenceGenome:
sequence_bits: bitarray
sequence_str: str # Stocke la séquence en chaîne
CARTE_INVERSE = CARTE_INVERSE
CARTE_NUCLEOTIDES = CARTE_NUCLEOTIDES
DECODE_TREE = DECODE_TREE
ASSOCI_COMB = ASSOCI_COMB
@classmethod
def from_string(cls, sequence: str):
bit_seq = bitarray()
bit_seq.encode(cls.CARTE_NUCLEOTIDES, sequence)
return cls(sequence_bits=bit_seq, sequence_str=sequence)
@classmethod
def from_bits(cls, bit_seq: bitarray):
bit_seq_copy = bit_seq.copy()
# Utilise la méthode 'decode' de l'objet bitarray en lui passant l'arbre de décodage
sequence_str = ''.join(bit_seq_copy.decode(cls.DECODE_TREE))
return cls(sequence_bits=bit_seq, sequence_str=sequence_str)
def to_string(self) -> str:
return self.sequence_str
def __len__(self):
return len(self.sequence_bits) // 2
def _inverse(self) -> bitarray:
valeurs_décodées = self.sequence_bits.decode(self.CARTE_NUCLEOTIDES)
# Applique la transformation via ASSOCI_COMB
valeurs_transformées = self.sequence_bits.decode(self.ASSOCI_COMB)
# Reencode les symboles transformés en bits (2 bits chacun)
résultat = bitarray()
résultat.encode(self.CARTE_NUCLEOTIDES, valeurs_transformées)
# lecture inverse
résultat.reverse()
return résultat
#----------------------------------------------------------------- Classe traducteurADN ---------------------------------------------------#
class traducteurADN:
def __init__(self):
self.CARTE_NUCLEOTIDES = CARTE_NUCLEOTIDES
# Mapping complet des codons (chaîne -> entier sous format qui explicite l'écriture 5 bits)
self.codons = {
# alanine
'GCT': 0b00000, 'GCC': 0b00000, 'GCA': 0b00000, 'GCG': 0b00000,
# arginine
'CGT': 0b00001, 'CGC': 0b00001, 'CGA': 0b00001, 'CGG': 0b00001,
'AGA': 0b00001, 'AGG': 0b00001,
# asparagine
'AAT': 0b00010, 'AAC': 0b00010,
# acide aspartique
'GAT': 0b00011, 'GAC': 0b00011,
# cystéine
'TGT': 0b00100, 'TGC': 0b00100,
# glutamine
'CAA': 0b00101, 'CAG': 0b00101,
# acide glutamique
'GAA': 0b00110, 'GAG': 0b00110,
# glycine
'GGT': 0b00111, 'GGC': 0b00111, 'GGA': 0b00111, 'GGG': 0b00111,
# histidine
'CAT': 0b01000, 'CAC': 0b01000,
# isoleucine
'ATT': 0b01001, 'ATC': 0b01001, 'ATA': 0b01001,
# leucine
'TTA': 0b01010, 'TTG': 0b01010, 'CTT': 0b01010, 'CTC': 0b01010,
'CTA': 0b01010, 'CTG': 0b01010,
# lysine
'AAA': 0b01011, 'AAG': 0b01011,
# méthionine (start)
'ATG': 0b01100,
# phénylalanine
'TTT': 0b01101, 'TTC': 0b01101,
# proline
'CCT': 0b01110, 'CCC': 0b01110, 'CCA': 0b01110, 'CCG': 0b01110,
# sérine
'TCT': 0b01111, 'TCC': 0b01111, 'TCA': 0b01111, 'TCG': 0b01111,
'AGT': 0b01111, 'AGC': 0b01111,
# thréonine
'ACT': 0b10000, 'ACC': 0b10000, 'ACA': 0b10000, 'ACG': 0b10000,
# tryptophane
'TGG': 0b10001,
# tyrosine
'TAT': 0b10010, 'TAC': 0b10010,
# valine
'GTT': 0b10011, 'GTC': 0b10011, 'GTA': 0b10011, 'GTG': 0b10011,
# STOP codons
'TAG': 0b11111, 'TAA': 0b11111, 'TGA': 0b11111
}
# Mapping du code entier 5 bits -> lettre d'acide aminé
self.acides_aminés = {
0b00000: 'A', 0b00001: 'R', 0b00010: 'N', 0b00011: 'D',
0b00100: 'C', 0b00101: 'Q', 0b00110: 'E', 0b00111: 'G',
0b01000: 'H', 0b01001: 'I', 0b01010: 'L', 0b01011: 'K',
0b01100: 'M', 0b01101: 'F', 0b01110: 'P', 0b01111: 'S',
0b10000: 'T', 0b10001: 'W', 0b10010: 'Y', 0b10011: 'V',
0b11111: 'STOP'
}
self.association_canonique = associ_canonique(self.codons, self.acides_aminés, CARTE_NUCLEOTIDES)
self.table_normalisation_complete = table_norm(self.codons, self.acides_aminés, self.association_canonique, CARTE_NUCLEOTIDES)
self.asso_normalisation_codon = {}
for codon in self.codons:
lettre_aa = self.acides_aminés[self.codons[codon]]
if lettre_aa == 'STOP':
continue
bits = frozenbitarray("".join(CARTE_NUCLEOTIDES[nt].to01() for nt in codon))
self.asso_normalisation_codon[bits] = self.association_canonique[lettre_aa]
self.arbre_decodage_codon = arbre_decodage(self.association_canonique)
self.carte_encodage_acides_aminés = carte_encodage_acides(self.acides_aminés)
patterns_recognized = [key.to01() for key in self.asso_normalisation_codon.keys()]
# Le motif tente de se concorder soit avec un codon reconnu (6 bits) soit exactement 2 bits à sauter pour réessayer avec les 6 bits suivants.
motif = r'(?:(?P<codon>' + '|'.join(patterns_recognized) + r')|(?P<skip>[01]{2}))'
self._codon_regex = regex.compile(motif)
def traduire_sequence(self, genome: sequenceGenome) -> bitarray:
bit_str = genome.sequence_bits.to01()
# Utilisation de regex.sub qui va parcourir toute la chaîne.
# Pour chaque correspondance, la fonction _fonction_substitution sera appelée.
chaine_normalisee = self._codon_regex.sub(self._fonction_substitution, bit_str)
# Conversion de la chaîne normalisée en bitarray.
normalisé = bitarray(chaine_normalisee)
# on s'assure que la longueur est un multiple de 6 bits.
if len(normalisé) % 6:
normalisé = normalisé[:len(normalisé) - (len(normalisé) % 6)]
# Décodage des codons normalisés en lettre acides aminés.
liste_aa = normalisé.decode(self.arbre_decodage_codon)
# On filtre pour garder uniquement les acides aminés présents dans la carte.
liste_aa_valides = filter(self.carte_encodage_acides_aminés.__contains__, liste_aa)
# Encodage des lettres en un bitarray protéique (5 bits par acide aminé).
protéine = bitarray()
protéine.encode(self.carte_encodage_acides_aminés, liste_aa_valides)
return protéine
def traduire_sequence_bits(self, adn: bitarray) -> bitarray:
bit_str = adn.to01()
chaine_normalisee = self._codon_regex.sub(self._fonction_substitution, bit_str)
normalisé = bitarray(chaine_normalisee)
if len(normalisé) % 6:
normalisé = normalisé[:len(normalisé) - (len(normalisé) % 6)]
# décodage des codons normalisés en lettres d'acides aminés.
liste_aa = normalisé.decode(self.arbre_decodage_codon)
#filtrer pour ne garder que les acides aminés présents dans la carte.
liste_aa_valides = filter(self.carte_encodage_acides_aminés.__contains__, liste_aa)
# Encodage des lettres en un bitarray protéique (5 bits par acide aminé).
protéine = bitarray()
protéine.encode(self.carte_encodage_acides_aminés, liste_aa_valides)
return protéine
def _fonction_substitution(self, match):
# Si le groupe 'codon' est reconnu, on le remplace par sa forme canonique.
if match.group('codon'):
codon = match.group('codon')
canonique = self.asso_normalisation_codon[frozenbitarray(codon)]
return canonique.to01()
# Sinon, c'est le groupe à ignorer qui a été matché, on retourne une chaîne vide pour ignorer ces 2 bits.
return ''
def traduire_sequence_bits_v1(self, adn: bitarray) -> bitarray:
bits = adn.copy()
bits = bits[:len(bits) - (len(bits) % 6)] if len(bits) % 6 else bits
# Normalisation de chaque codon en utilisant la map de normalisation (boucle sur les codons)
normalisé = bitarray()
for i in range(0, len(bits), 6):
codon_bits = bits[i:i+6]
normalisé.extend(self.asso_normalisation_codon.get(frozenbitarray(codon_bits), codon_bits))
# Décodage des codons normalisés en lettres d'acides aminés
liste_aa = normalisé.decode(self.arbre_decodage_codon)
liste_aa_valides = filter(self.carte_encodage_acides_aminés.__contains__, liste_aa)
# Encodage des lettres en un bitarray protéique (5 bits par acide aminé)
protéine = bitarray()
protéine.encode(self.carte_encodage_acides_aminés, liste_aa_valides)
return protéine
def proteine_to_string(self, protéine: bitarray) -> str:
return "".join(
self.acides_aminés.get(int(protéine[i:i+5].to01(), 2), '?')
for i in range(0, len(protéine), 5)
)
# ----------------------------------------------------------------------------Test -----------------------------------------------
genome = sequenceGenome.from_string("ATGATGGTGACGTAT")
traducteur = traducteurADN()
protéine = traducteur.traduire_sequence(genome)
print("séquence adn:", genome.to_string())
print("Sequence protéique bitarray traduite d'une ADN chaine:", protéine)
print("protéine traduite:", traducteur.proteine_to_string(protéine))
# Test avec une séquence ADN codée en bitarray
adn_test_bits = bitarray("000111000111110111001011010001") # Correspond à ATGATGGTGACGTAT
protéine_test = traducteur.traduire_sequence_bits(adn_test_bits)
print("\n\nSéquence ADN introduite brute en bits:", adn_test_bits)
print("Séquence ADN chargée en bits:", genome.sequence_bits)
print("Séquence protéique en bits:", protéine_test)séquence adn: ATGATGGTGACGTAT
Sequence protéique bitarray traduite d'une ADN chaine: bitarray('0110001100100111000010010')
protéine traduite: MMVTY
Séquence ADN introduite brute en bits: bitarray(‘000111000111110111001011010001’)
Séquence ADN chargée en bits: bitarray(‘000111000111110111001011010001’)
Séquence protéique en bits: bitarray(‘0110001100100111000010010’)
##2. Lecture d’un génome et recherche de regions codantes.
@dataclass
class cadresLecture:
genome: object
# Codon de départ (ATG) en 6 bits
CODON_START = frozenbitarray('000111')
# Codons d'arrêt (TGA, TAG, TAA) en 6 bits
CODON_STOP = {frozenbitarray('010011'), frozenbitarray('010000'), frozenbitarray('011100')}
# Dictionnaire de conversion inverse des 2 bits en nucléotide
CARTE_INVERSE = {"00": "A", "01": "T", "10": "C", "11": "G"}
def obtenir_cadre(self, sens, decalage):
# Récupère la séquence de codons (6 bits par codon) pour un cadre de lecture donné
# Si sens==1, on utilise la séquence normale, sinon la séquence inverse
seq = self.genome.sequence_bits if sens == 1 else self.genome._inverse()
debut = decalage * 2 # Chaque nucléotide = 2 bits
# Extraction vectorisée de tous les codons dans une liste
return [seq[i:i+6] for i in range(debut, len(seq)-5, 6)]
def tous_les_cadres(self):
# Retourne un dictionnaire contenant les 6 cadres de lecture
return {(sens, decalage): self.obtenir_cadre(sens, decalage)
for sens in [1, -1] for decalage in range(3)}
def identifier_regions_codantes_vectorise(self, cadre):
if not cadre:
return []
# Conversion vectorielle : créer un tableau numpy contenant la valeur entière de chaque codon
# Chaque codon est converti en entier via sa représentation binaire sur 6 bits
valeurs_codons = np.array([int(codon.to01(), 2) for codon in cadre], dtype=np.uint8)
# Valeur entière du codon de départ (start)
val_start = int(self.CODON_START.to01(), 2)
# Ensemble des valeurs entières pour les codons d'arrêt (stop)
ensemble_stop = np.array([int(c.to01(), 2) for c in self.CODON_STOP], dtype=np.uint8)
# Indices où le codon est le codon start
indices_start = np.where(valeurs_codons == val_start)[0]
# Indices où le codon est un codon stop
indices_stop = np.where(np.isin(valeurs_codons, ensemble_stop))[0]
if indices_start.size == 0 or indices_stop.size == 0:
return []
# Pour chaque indice de début, obtenir l'indice du premier codon stop qui le suit (vectorisé)
positions = np.searchsorted(indices_stop, indices_start, side='right')
# Masque : positions valides (position < nombre d'indices stop)
masque_valide = positions < indices_stop.size
if not np.any(masque_valide):
return []
debuts_valides = indices_start[masque_valide]
positions_valides = positions[masque_valide]
stops_associes = indices_stop[positions_valides]
# Calculer la longueur de chaque région candidate
longueurs = stops_associes - debuts_valides + 1
# Filtrer les régions dont la longueur est d'au moins 100 codons
masque_regions = longueurs >= 3 # à changer a la valeur 3 ici pour les tests
debuts_final = debuts_valides[masque_regions]
stops_final = stops_associes[masque_regions]
# Extraction vectorisée des régions codantes depuis le cadre
# (On utilise ici une liste car le découpage d'une liste d'objets n'est pas aisément vectorisable)
régions = [cadre[i:j] for i, j in zip(debuts_final, stops_final)]
return régions
def toutes_les_regions_codantes(self):
# Retourne un dictionnaire avec, pour chaque cadre, la liste des régions codantes détectées
cadres = self.tous_les_cadres()
return {cle: self.identifier_regions_codantes_vectorise(cadre) for cle, cadre in cadres.items()}
def debug_affichage_codons(self):
# Affiche, pour chaque cadre, la séquence de codons sous forme de nucléotides
print("\n=== Debug: Affichage des cadres sous forme de nucléotides ===")
tous_les_cadres = self.tous_les_cadres()
for cle, regions in tous_les_cadres.items():
sequences = [
"".join(self.CARTE_INVERSE[r[i:i+2].to01()] for i in range(0, len(r), 2))
for r in regions
]
print(f"cadre {cle} : {sequences}")
def proportion_codons_potentiels(self, frame_input):
#Calcule la proportion de codons dans chaque cadre de lecture qui sont potentiellement exprimés (c'est-à-dire qui font partie d'une région codante,
#sauf le codon STOP final de chaque région).
#Retourne un dictionnaire dont les clés sont les tuples (sens, decalage) et les valeurs la proportion de codons exprimés dans le cadre.
# Si cadre est une clé (tuple), extraire le cadre correspondant.
if isinstance(frame_input, tuple):
cadres = self.tous_les_cadres()
if frame_input not in cadres:
raise ValueError(f"Le cadre de lecture {frame_input} n'existe pas.")
cadre = cadres[frame_input]
else:
cadre = frame_input
total_codons = len(cadre)
if total_codons == 0:
return 0.0
# Extraire les régions codantes pour ce cadre.
region = self.identifier_regions_codantes_vectorise(cadre)[0]
# Pour chaque région, le nombre de codons exprimés est: longueur de la région. ( ou normalement longueur de la région - 1 ? car on exclut le codon STOP final... )
codons_potentiels = len(region) if region else 0
return codons_potentiels / total_codons
# ---------------------------------------------------------------------------------------------TEST ----------------------------------------------
genome = sequenceGenome.from_string("GAGTTATGCAGTGGTAGTATGA")
analyseur = cadresLecture(genome)
#analyseur.debug_affichage_codons()
cadres = analyseur.tous_les_cadres()
for cle, cadre in cadres.items():
print(f"{cle} : {cadre}")
regions_codantes = analyseur.toutes_les_regions_codantes()
for cle, regions in regions_codantes.items():
if regions:
print(f"régions codantes pour {cle} : {regions}")(1, 0) : [bitarray('110011'), bitarray('010100'), bitarray('011110'), bitarray('001101'), bitarray('111101'), bitarray('001101'), bitarray('000111')]
(1, 1) : [bitarray('001101'), bitarray('010001'), bitarray('111000'), bitarray('110111'), bitarray('110100'), bitarray('110100'), bitarray('011100')]
(1, 2) : [bitarray('110101'), bitarray('000111'), bitarray('100011'), bitarray('011111'), bitarray('010011'), bitarray('010001')]
(-1, 0) : [bitarray('001101'), bitarray('000111'), bitarray('000111'), bitarray('110111'), bitarray('001011'), bitarray('010001'), bitarray('011100')]
(-1, 1) : [bitarray('110100'), bitarray('011100'), bitarray('011111'), bitarray('011100'), bitarray('101101'), bitarray('000101'), bitarray('110011')]
(-1, 2) : [bitarray('010001'), bitarray('110001'), bitarray('111101'), bitarray('110010'), bitarray('110100'), bitarray('010111')]
régions codantes pour (1, 2) : [[bitarray('000111'), bitarray('100011'), bitarray('011111')]]
régions codantes pour (-1, 0) : [[bitarray('000111'), bitarray('000111'), bitarray('110111'), bitarray('001011'), bitarray('010001')], [bitarray('000111'), bitarray('110111'), bitarray('001011'), bitarray('010001')]]
#test
for codon in regions_codantes[(-1,0)][0]:
seq_codon = sequenceGenome.from_bits(codon)
print(f"régions codantes pour :{seq_codon.sequence_str}")régions codantes pour :ATG
régions codantes pour :ATG
régions codantes pour :GTG
régions codantes pour :ACG
régions codantes pour :TAT
analyseur.tous_les_cadres(){(1, 0): [bitarray('110011'),
bitarray('010100'),
bitarray('011110'),
bitarray('001101'),
bitarray('111101'),
bitarray('001101'),
bitarray('000111')],
(1, 1): [bitarray('001101'),
bitarray('010001'),
bitarray('111000'),
bitarray('110111'),
bitarray('110100'),
bitarray('110100'),
bitarray('011100')],
(1, 2): [bitarray('110101'),
bitarray('000111'),
bitarray('100011'),
bitarray('011111'),
bitarray('010011'),
bitarray('010001')],
(-1, 0): [bitarray('001101'),
bitarray('000111'),
bitarray('000111'),
bitarray('110111'),
bitarray('001011'),
bitarray('010001'),
bitarray('011100')],
(-1, 1): [bitarray('110100'),
bitarray('011100'),
bitarray('011111'),
bitarray('011100'),
bitarray('101101'),
bitarray('000101'),
bitarray('110011')],
(-1, 2): [bitarray('010001'),
bitarray('110001'),
bitarray('111101'),
bitarray('110010'),
bitarray('110100'),
bitarray('010111')]}
analyseur.proportion_codons_potentiels((-1,0))0.7142857142857143
3 Lecture des fichiers FASTA
3.1.1 Charger un fichier FASTA contenant un génome: GCA_046529535.1.fasta.gz
import os
import gzip
import requests
import re
import shutil
# Dictionnaire de conversion nucléotides à des chaine de caractères qui exolplicitent la representations bits
CARTE_NUC_LEC = {
'A': '00',
'T': '01',
'C': '10',
'G': '11'
}
# Création du mapping d'encodage pour bitarray.encode : lettre --> bitarray
CARTE_ENCODAGE = {lettre: bitarray(bits) for lettre, bits in CARTE_NUC_LEC.items()}
# Création d'un arbre de décodage pour bitarray.decode à partir du mapping d'encodage
ARBRE_DECODAGE = decodetree(CARTE_ENCODAGE)
# ------------------------------------------------------------------------------------ Classe LecteurFASTA ----------------------------------------------------------------
class LecteurFASTA:
def __init__(self, chemin_fichier):
self.chemin_fichier = chemin_fichier
self.nom_fichier = ""
self.sequence_bits = bitarray()
self.sequence_str = ""
def telecharger_fichier(self):
# si le chemin est une url
if self.chemin_fichier.startswith("http"):
print(f"Téléchargement de {self.chemin_fichier}")
reponse = requests.get(self.chemin_fichier)
if reponse.status_code == 200:
disposition = re.search(r'filename="(.+)"', reponse.headers.get("Content-Disposition", ""))
if disposition:
self.nom_fichier = disposition.group(1)
else:
self.nom_fichier = self.chemin_fichier.split("/")[-1]
if not self.nom_fichier.endswith(".gz"):
self.nom_fichier += ".fasta.gz"
with open(self.nom_fichier, "wb") as f:
f.write(reponse.content)
self.chemin_fichier = self.nom_fichier
print(f"Téléchargement terminé : {self.nom_fichier}")
else:
raise ValueError(f"Erreur du téléchargement : {reponse.status_code}")
def est_fichier_gzip(self):
with open(self.chemin_fichier, 'rb') as f:
signature = f.read(2)
return signature == b'\x1f\x8b'
def decompresser_fichier(self):
# Décompresse le fichier gzip s'il l'est
if self.chemin_fichier.endswith('.gz') and self.est_fichier_gzip():
fichier_decompresse = self.chemin_fichier.replace('.gz', '.fasta')
with gzip.open(self.chemin_fichier, 'rb') as f_in, open(fichier_decompresse, 'wb') as f_out:
shutil.copyfileobj(f_in, f_out)
self.chemin_fichier = fichier_decompresse
print(f"Décompression terminée : {self.chemin_fichier}")
else:
print(f"Le fichier {self.chemin_fichier} ne semble pas être un gzip, lecture directe.")
def lire_fasta_adn(self):
self.telecharger_fichier()
self.decompresser_fichier()
if os.path.exists(self.chemin_fichier):
with open(self.chemin_fichier, 'r', encoding='utf-8') as f:
# Lit les lignes et filtre celles commençant par ">"
lignes = f.read().splitlines()
séquence = "".join(l for l in lignes if not l.startswith(">"))
# Définir l'ensemble des caractères valides
valid_chars = set("ACGT") # Ex: Acides aminés valides
# Filtrer les caractères invalides en utilisant filter()
filtered_data = "".join(filter(valid_chars.__contains__, séquence))
self.sequence_str = filtered_data
def lire_fasta_prot(self):
self.telecharger_fichier()
self.decompresser_fichier()
if os.path.exists(self.chemin_fichier):
with open(self.chemin_fichier, 'r', encoding='utf-8') as f:
# Lit les lignes et filtre celles commençant par ">"
lignes = f.read().splitlines()
séquence = "".join(l for l in lignes if not l.startswith(">"))
# Définir l'ensemble des caractères valides
valid_chars = set("ACHMTRQIFWNELPYDGKSV") # Ex: Acides aminés valides
# Filtrer les caractères invalides en utilisant filter()
filtered_data = "".join(filter(valid_chars.__contains__, séquence))
self.sequence_str = filtered_data
def obtenir_sequence_bits_adn(self):
self.sequence_bits = bitarray()
self.sequence_bits.encode(CARTE_ENCODAGE, self.sequence_str)
return self.sequence_bits
#--------------------------------------------------------------------------------------------------Usage---------------------------------------
lecteur_adn = LecteurFASTA("https://www.ebi.ac.uk/ena/browser/api/fasta/GCA_046529535.1?download=true&gzip=true")
lecteur_adn.lire_fasta_adn()
print("Séquence :", lecteur_adn.sequence_str)Téléchargement de https://www.ebi.ac.uk/ena/browser/api/fasta/GCA_046529535.1?download=true&gzip=true
Téléchargement terminé : GCA_046529535.1?download=true&gzip=true.fasta.gz
Décompression terminée : GCA_046529535.1?download=true&gzip=true.fasta.fasta
Séquence : ATCTTTTTCGGCTTTTTTT ...
sequence_adn_bits = lecteur_adn.obtenir_sequence_bits_adn()
# affichage de la séquence en bits
print(f"Séquence ADN en bits (échantillon) : {sequence_adn_bits[:50]}")
# affichage de la séquence ADN au format chaîne de caractères
sequence_adn_str = lecteur_adn.sequence_str
print(f"Séquence ADN en chaine de caractères (échantillon) : {sequence_adn_str[:50]}")Séquence ADN en bits (échantillon) : bitarray('00011001010101011011111001010101010101001101000110')
Séquence ADN en chaine de caractères (échantillon) : ATCTTTTTCGGCTTTTTTTAGTATCCACAGAGGTTATCGACAACATTTTC
Genome_=sequenceGenome.from_bits(sequence_adn_bits)
print(Genome_.sequence_bits[:50])
print(Genome_.sequence_str[:50])bitarray('00011001010101011011111001010101010101001101000110')
ATCTTTTTCGGCTTTTTTTAGTATCCACAGAGGTTATCGACAACATTTTC
3.1.2 Stocker une séquence de protéine de proteines.
Le meme code précédent peut etre adapté pour stocker une séquence de proteines.
# ------------------------------------------------------------- classe pour stocker une séquence protéique --------------------------------------------
@dataclass
class sequenceProteine:
sequence: bitarray
sequence_str: str
# dictionnaire de conversion des acides aminés
CARTE_INVERSE_AA = {
frozenbitarray('00000'): 'A', frozenbitarray('00001'): 'R', frozenbitarray('00010'): 'N', frozenbitarray('00011'): 'D',
frozenbitarray('00100'): 'C', frozenbitarray('00101'): 'Q', frozenbitarray('00110'): 'E', frozenbitarray('00111'): 'G',
frozenbitarray('01000'): 'H', frozenbitarray('01001'): 'I', frozenbitarray('01010'): 'L', frozenbitarray('01011'): 'K',
frozenbitarray('01100'): 'M', frozenbitarray('01101'): 'F', frozenbitarray('01110'): 'P', frozenbitarray('01111'): 'S',
frozenbitarray('10000'): 'T', frozenbitarray('10001'): 'W', frozenbitarray('10010'): 'Y', frozenbitarray('10011'): 'V',
frozenbitarray('11111'): 'STOP'
}
CARTE_AMINO_ACIDES = {v: k for k, v in CARTE_INVERSE_AA.items()}
@classmethod
def from_string(cls, sequence: str):
bit_seq = bitarray()
valid_sequence = filter(cls.CARTE_AMINO_ACIDES.__contains__, sequence)
bit_seq.encode(cls.CARTE_AMINO_ACIDES, valid_sequence)
return cls(sequence=bit_seq, sequence_str="".join(valid_sequence))
def load_string(self) -> str:
return self.sequence_str
def __len__(self):
return len(self.sequence) // 5
def sequence_to_string(self, proteine: bitarray) -> str:
return "".join(proteine.decode(self.CARTE_AMINO_ACIDES))
# test
proteine = sequenceProteine.from_string("MMVTY")
print("séquence protéique chargée:", proteine.load_string())
print("séquence binaire:", proteine.sequence)
print("conversion test:", proteine.sequence_to_string(proteine.sequence))séquence protéique chargée:
séquence binaire: bitarray('0110001100100111000010010')
conversion test: MMVTY
3.1.2 Lecteur de fichier FASTA contenant une séquence protéique
Le lecteur FASTA peut-etre modifié pour encoder aussi des sequence proteines mais on va juste coimbiner les fonctionnalité des codes précédents :
lecteur_prot = LecteurFASTA("https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz")
lecteur_prot.lire_fasta_prot()Téléchargement de https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz
Téléchargement terminé : uniprot_sprot.fasta.gz
Décompression terminée : uniprot_sprot.fasta.fasta
lecteur_prot.sequence_str[:50]'MAFSAEDVLKEYDRRRRMEALLLSLYYPNDRKLLDYKEWSPPRVQVECPK'
str_prot = lecteur_prot.sequence_str
sequence_prot = sequenceProteine.from_string(str_prot)sequence_prot.sequence[:50]bitarray('01100000000110101111000000011000011100110101001011')
3.2 Rechrcher les meilleurs cadres (ceux qui sont potentiellement exprimés)
Évalue chaque cadre en prenant en compte le nombre total et la longueur des régions codantes (pas seulement la plus longue comme le code précédent). Puis classe les cadres de lecture en fonction du nombre de séquences codantes valides qu’ils contiennent et détermine le meilleur cadre de lecture global (pas seulement une longue région, mais le cadre ayant la plus grande pertinence biologique). Il suppose et se base sur les conditions basiques suivantes :
- Un cadre contenant beaucoup de régions codantes distinctes est plus probable d’être le bon.
- Seul le total des longueurs est pris en compte, pas le nombre de fragments.
# Classe pour analyser et identifier les cadres de lecture les plus probables
class AnalyseCadres:
def __init__(self, genome: bitarray):
# initialise l’analyse des cadres de lecture d’une séquence génomique
# on suppose que 'cadresLecture' a été optimisé pour manipuler des bitarrays directement
self.genome = genome
self.analyseur = cadresLecture(self.genome)
def scorer_cadres(self):
# évalue chaque cadre en fonction du nombre et de la taille des régions codantes
scores = {}
regions_codantes = self.analyseur.toutes_les_regions_codantes()
for cadre, regions in regions_codantes.items():
# 'regions' est une liste de régions; chaque région est une liste de codons sous forme bitarray
# on calcule simplement la somme du nombre de codons (len(region)) pour chaque région
score = sum(len(region) for region in regions)
scores[cadre] = score
return scores
def cadre_le_plus_probable(self):
# retourne le cadre de lecture avec le plus haut score
scores = self.scorer_cadres()
if not scores:
return None, 0
meilleur_cadre = max(scores, key=scores.get) # cadre avec le plus grand score
return meilleur_cadre, scores[meilleur_cadre]
def afficher_resultats(self):
# affiche les cadres de lecture triés par probabilité
scores = self.scorer_cadres()
cadres_tries = sorted(scores.items(), key=lambda x: x[1], reverse=True)
print("\nCadres de lecture triés par score :")
for cadre, score in cadres_tries:
print(f"Cadre {cadre} ; Score : {score}")
# utilisation ( pourle test changer la limite à 3 : identifier_regions_codantes() de la classe genome dans 2. Lecture d'un genome )
genome = sequenceGenome.from_string("GAGTTATGCAGTGGTAGTATGA")
analyse = AnalyseCadres(genome)
# affichage des résultats
analyse.afficher_resultats()
# meilleur cadre de lecture
meilleur_cadre, score = analyse.cadre_le_plus_probable()
print(f"\nCadre de lecture le plus probable : {meilleur_cadre} avec un score de {score}")Cadres de lecture triés par score :
Cadre (-1, 0) ; Score : 9
Cadre (1, 2) ; Score : 3
Cadre (1, 0) ; Score : 0
Cadre (1, 1) ; Score : 0
Cadre (-1, 1) ; Score : 0
Cadre (-1, 2) ; Score : 0
Cadre de lecture le plus probable : (-1, 0) avec un score de 9
from itertools import chain
# Initialisation de l'analyseur avec un ADN en bitarray
genome = sequenceGenome.from_string("GAGTTATGCAGTGGTAGTATGA")
analyseurt = AnalyseCadres(genome)
# Récupération du cadre détecté
regions_codantes = analyseurt.analyseur.toutes_les_regions_codantes()
meilleur_cadre, _ = analyseurt.cadre_le_plus_probable()
print(f"Meilleur cadre détecté: {meilleur_cadre}")
# affichage des régions codantes
region_probable = regions_codantes[meilleur_cadre]
region_probableMeilleur cadre détecté: (-1, 0)
[[bitarray('000111'),
bitarray('000111'),
bitarray('110111'),
bitarray('001011'),
bitarray('010001')],
[bitarray('000111'),
bitarray('110111'),
bitarray('001011'),
bitarray('010001')]]
Concaténer toutes les régions du meilleur cadre :
# Aplatir la liste imbriquée
liste_aplatie = list(chain.from_iterable(region_probable))
# Fusionner en un seul bitarray en utilisant sum (bitarray supporte la concaténation)
bitarray_fusionné = sum(liste_aplatie, bitarray())
print(bitarray_fusionné)bitarray('000111000111110111001011010001000111110111001011010001')
Concaténer chaque région du meilleur cadre:
# Fusionner chaque sous-liste en un seul bitarray en utilisant map() et sum()
liste_fusionnée = list(map(lambda sous_liste: sum(sous_liste, bitarray()), region_probable))
# Afficher les listes fusionnées
print(liste_fusionnée)[bitarray('000111000111110111001011010001'), bitarray('000111110111001011010001')]
Concaténer la region en position 1 du cadre :
position_1 = sum(region_probable[0], bitarray())
position_1bitarray('000111000111110111001011010001')
#vérification
seq_position_1 = sequenceGenome.from_bits(position_1)
seq_position_1.sequence_str'ATGATGGTGACGTAT'
4. Applications
Détection des régions codantes sur un génome complet.
Précédemment, on a chargé la sequence adn Genome_ à partir du fichier GCA_046529535.1.fasta.gz et la séquence proteine sequence_prot à partir du fichier uniprot_sprot.fasta.fasta.
On va maintenant essayer de trouver les regions de GCA_046529535.1 susceptibles d’exprimer des proteines de uniprot_sprot.fasta.fasta.
analyseur_brute = AnalyseCadres(Genome_)
meilleur_cadre_brute, score_brute = analyseur_brute.cadre_le_plus_probable()
print(f"\nCadre de lecture le plus probable : {meilleur_cadre_brute} avec un score de {score_brute}")Cadre de lecture le plus probable : (1, 1) avec un score de 1998173
analyseur_brute.analyseur.proportion_codons_potentiels((1,1))0.00035432523468440386
length_brute = len(analyseur_brute.analyseur.tous_les_cadres()[meilleur_cadre_brute])
length_brute1337754
4.1 Traduire l’adn
CODONS_LIST = [
# Alanine
'GCT', 'GCC', 'GCA', 'GCG',
# Arginine
'CGT', 'CGC', 'CGA', 'CGG', 'AGA', 'AGG',
# Asparagine
'AAT', 'AAC',
# Aspartic acid
'GAT', 'GAC',
# Cysteine
'TGT', 'TGC',
# Glutamine
'CAA', 'CAG',
# Glutamic acid
'GAA', 'GAG',
# Glycine
'GGT', 'GGC', 'GGA', 'GGG',
# Histidine
'CAT', 'CAC',
# Isoleucine
'ATT', 'ATC', 'ATA',
# Leucine
'TTA', 'TTG', 'CTT', 'CTC', 'CTA', 'CTG',
# Lysine
'AAA', 'AAG',
# Methionine (start)
'ATG',
# Phenylalanine
'TTT', 'TTC',
# Proline
'CCT', 'CCC', 'CCA', 'CCG',
# Serine
'TCT', 'TCC', 'TCA', 'TCG', 'AGT', 'AGC',
# Threonine
'ACT', 'ACC', 'ACA', 'ACG',
# Tryptophan
'TGG',
# Tyrosine
'TAT', 'TAC',
# Valine
'GTT', 'GTC', 'GTA', 'GTG',
# STOP codons
'TAG', 'TAA', 'TGA'
]adn_arr = np.array(list(Genome_.sequence_str))
#sequence_adn_bits.encode(CODONS, Genome_.sequence_str)windows = np.lib.stride_tricks.sliding_window_view(adn_arr, 3)
# Reconstruction vectorisée des codons (chaînes de 3 caractères)
codons = np.char.add(np.char.add(windows[:, 0], windows[:, 1]), windows[:, 2])
# Création d'un tableau contenant les codons valides et leurs inverses
valid_array = np.array(list(CODONS_LIST))
valid_inv = np.array([s[::-1] for s in CODONS_LIST])
#valid_set = np.concatenate([valid_array, valid_inv])
valid_inv[:50]array(['TCG', 'CCG', 'ACG', 'GCG', 'TGC', 'CGC', 'AGC', 'GGC', 'AGA',
'GGA', 'TAA', 'CAA', 'TAG', 'CAG', 'TGT', 'CGT', 'AAC', 'GAC',
'AAG', 'GAG', 'TGG', 'CGG', 'AGG', 'GGG', 'TAC', 'CAC', 'TTA',
'CTA', 'ATA', 'ATT', 'GTT', 'TTC', 'CTC', 'ATC', 'GTC', 'AAA',
'GAA', 'GTA', 'TTT', 'CTT', 'TCC', 'CCC', 'ACC', 'GCC', 'TCT',
'CCT', 'ACT', 'GCT', 'TGA', 'CGA'], dtype='<U3')
# Test vectorisé de la validité de chaque codon
is_valid_direct = np.isin(codons, valid_array)
is_valid_inv = np.isin(codons, valid_inv)
# Récupération des indices (dans codons) des fenêtres valides
indices_direct = np.nonzero(is_valid_direct)[0]
indices_inv = np.nonzero(is_valid_inv)[0]
# Sélection non-chevauchante des codons valides (greedy)
if indices_direct.size == 0:
sequence_valide_direct = ""
else:
chosen = [indices_direct[0]]
for idx in indices_direct[1:]:
if idx >= chosen[-1] + 3:
chosen.append(idx)
# Reconstruction de la chaîne résultat à partir des codons choisis
sequence_valide_direct = "".join(codons[chosen])
if indices_inv.size == 0:
sequence_valide_inv = ""
else:
chosen = [indices_inv[0]]
for idx in indices_inv[1:]:
if idx >= chosen[-1] + 3:
chosen.append(idx)
# Reconstruction de la chaîne résultat à partir des codons choisis
sequence_valide_inv= "".join(codons[chosen])
print(len(sequence_valide_inv))
print(len(sequence_valide_direct))4013262
4013262
trqducteur_ = traducteurADN()Étapes de filtrage des codons :
On souhaite ne conserver dans la séquence d’ADN que les codons qui codent pour des protéines. Cette étape pourrait être biologiquement non pertinente, rédondante ou logiquement superflue, car en réalité les condons des listes des régions codantes (et non pas des cadres de lectures) sont déjà des codons identifiés valides. Je m’en sert comme double vérification.
L’objectif est d’obtenir des séquences de protéines traduites (après filtrage basé sur un dictionnaire de codons) en ignorant les codons qui ne correspondent pas à des protéines répertoriées, puis comparer ces séquences à celles d’une base de données protéique et identifier des éventuelles correspondances.
Genome_d = sequenceGenome.from_string(sequence_valide_direct)
Genome_i = sequenceGenome.from_string(sequence_valide_inv)
#sequence_direct_to_prot_bits = traducteur_adn.traduire_sequence(Genome_d)
#sequence_inv_to_prot_bits = traducteur_adn.traduire_sequence(Genome_i)
#sequence_adn_bits.encode(CODONS, Genome_.sequence_str)Genome_d.sequence_bits[:50]bitarray('00011001010101011011111001010101010101001101000110')
analyseur_d = AnalyseCadres(Genome_d)
meilleur_cadre_d, score_d = analyseur_d.cadre_le_plus_probable()
print(f"\nCadre de lecture le plus probable après filtrage de chaine en lecture directe : {meilleur_cadre_d} avec un score de {score_d}")
analyseur_i = AnalyseCadres(Genome_i)
meilleur_cadre_i, score_i = analyseur_i.cadre_le_plus_probable()
print(f"\nCadre le plus probable après filtrage de chaine en lecture inverse: {meilleur_cadre_i} avec un score de {score_i}")Cadre de lecture le plus probable après filtrage de chaine en lecture directe : (1, 1) avec un score de 1998173
Cadre le plus probable après filtrage de chaine en lecture inverse: (1, 1) avec un score de 1998173
regions_codantes_d = analyseur_d.analyseur.toutes_les_regions_codantes()
regions_codantes_i = analyseur_d.analyseur.toutes_les_regions_codantes()regions_codantes_d == regions_codantes_iTrue
regions_exprimées = regions_codantes_d[meilleur_cadre_d]
# Fusionner chaque sous-liste en un seul bitarray en utilisant map() et sum()
liste_regions_fusionnée = list(map(lambda sl: sum(sl, bitarray()), regions_exprimées))
# Afficher un echantillon des listes fusionnées
liste_regions_fusionnée[0][:50]bitarray('00011111010000111011101000010101111001100101010101')
nb_codons = len(regions_exprimées[0])
proportions_codons = nb_codons/length_brute if nb_codons else 0
print(proportions_codons)0.00035432523468440386
region_potentielle_bits_filtrée = trqducteur_.traduire_sequence_bits(liste_regions_fusionnée[0])
regions_potentielles_traduites_en_proteine = trqducteur_.traduire_sequence_bits(region_potentielle_bits_filtrée)regions_potentielles_traduites_en_proteine[:50]bitarray('01111011110000001001010100010010011000011001100011')
len(regions_potentielles_traduites_en_proteine)1930
len(sequence_prot.sequence)1038064435
def trouver_correspondances_parfaites(segment_petit, segment_grand):
# Recherche toutes les positions où le segment petit apparaît dans le segment grand.
# Utilise la méthode search() de bitarray pour obtenir la liste des indices de début des correspondances.
positions = list(segment_grand.search(segment_petit))
return positions
# Trouver toutes les correspondances du segment petit dans le segment grand
positions_correspondances = trouver_correspondances_parfaites(regions_potentielles_traduites_en_proteine, sequence_prot.sequence)
# Afficher les indices où le segment petit est trouvé dans le segment grand
if positions_correspondances:
for indice in positions_correspondances:
print(f"Correspondance trouvée à l'indice : {indice}")
else:
print("Aucune correspondance parfaite trouvée de la meilleure region codante concaténées de GCA_046529535.1 avec la base uniprot_sprot.")Aucune correspondance parfaite trouvée de la meilleure region codante concaténée concaténées de GCA_046529535.1 avec la base uniprot_sprot.
En meme temps, devrait-on vraiment s’attendre à avoir une correspondance parafaite étant donné que les région codantes ont été concaténées ? J’ai utilisé ces deux bases de données comme l’exemple du projet sans vérification qu’ils ont une quelquonque connexion biologique ou que l’une devrait coder pour l’autre. Dans la vrai vie on ne sait pas quand on fait des tests, on veut justement le savoir. 🙄
Recherche déterministe des similarités :
Le code suivant detecte des régions similaires entre deux séquences binaires en les découpant en petits blocs:
-
Il convertit des bitarrays en tableaux numpy en deux dimensions, où chaque ligne correspond à un bloc de 5 bits.
-
Chaque bloc est ensuite transformé en une valeur entière entre 0 et 31 (pas très glamour étant et fait perdre les tableau de bitarray leur interet au final), ce qui facilite la comparaison.
-
On commence par identifier dans la séquence large toutes les positions où apparaît le premier bloc de la petite séquence. Pour chaque position candidate, une fonction compilée avec numba (pour avoir un temps d’exécution moins important) parcourt les deux séquences en comptant les blocs correspondants en permettant qu’un nombre de blocs ne concorde pas, ici un max de 3 blocs. Si le nombre de correspondances arrive à un certain nombre disons 50 dans ce cas, la région est sauvegardée et rajoutée aux regions considérées similaires ainsi que l’indice du début dans la grande séquence, nombre de correspondances et pourcentage de similarité.
import time
from numba import njit
#Convertir un bitarray en tableau numpy de blocs d'entiers où chaque bloc représente une valeur 0-31
def en_blocs(bitarr, taille_bloc=5):
bits = np.unpackbits(np.frombuffer(bitarr.tobytes(), dtype=np.uint8))[:len(bitarr)]
return bits.reshape(-1, taille_bloc)
def blocs_en_entiers(blocs):
poids = 2 ** np.arange(blocs.shape[1]-1, -1, -1, dtype=np.uint32)
return blocs.dot(poids)
#On cherche à enchaîner un alignement à partir d'une position candidate
@njit
def trouver_correspondance(grand, petit, i_0, max_ecart, min_corr):
n_grand = len(grand)
n_petit = len(petit)
i = i_0
j = 0
ecart = 0
nb_corr = 0
while i < n_grand and j < n_petit:
if grand[i] == petit[j]:
nb_corr += 1
ecart = 0 # Réinitialisation de l’écart si correspondance trouvée
j += 1
i += 1
else:
ecart += 1
i += 1 # On saute ce bloc dans le grand bitarray
if ecart > max_ecart:
break # Trop d'écarts consécutifs, abandonner cette région
if nb_corr >= min_corr:
return nb_corr
return nb_corr
# Fonction principale qui recherche les régions candidates et retourne les résultats
def regions_correspondantes(grand_bitarray, petit_bitarray, taille_bloc=5,
min_corr=50, max_ecart=3):
# Conversion des bitarrays en tableaux d'entiers
blocs_grand = en_blocs(grand_bitarray, taille_bloc)
blocs_petit = en_blocs(petit_bitarray, taille_bloc)
grand_entiers = blocs_en_entiers(blocs_grand)
petit_entiers = blocs_en_entiers(blocs_petit)
# Recherche des indices où le premier bloc du petit est présent dans le grand
indices_candidats = np.where(grand_entiers == petit_entiers[0])[0]
regions_s = []
nb_blocs_petit = len(petit_entiers)
for indice in indices_candidats:
correspondances = trouver_correspondance(grand_entiers, petit_entiers, indice, max_ecart, min_corr)
if correspondances >= min_corr:
pourcentage = correspondances / nb_blocs_petit
regions_s.append((indice * taille_bloc, correspondances, pourcentage))
return regions_s
#----------------------------------------------------------------TEST avec suivi du temps pour voir estimer la compléxité empiriquement -----------------------------
debut_temps = time.time()
regions = regions_correspondantes(sequence_prot.sequence, regions_potentielles_traduites_en_proteine, taille_bloc=5,
min_corr=10, max_ecart=6)
fin_temps = time.time()
# Affichage des résultats
if regions:
for position, nb_match, pourcentage in regions:
print(f"Région candidate trouvée à la position : {position} bits (Blocs correspondants : {nb_match}, "
f"Similarité : {pourcentage*100:.2f}%)")
else:
print("Aucune région candidate trouvée.")
print(f"Temps d'exécution : {fin_temps - debut_temps:.6f} sec")Région candidate trouvée à la position : 802890 bits (Blocs correspondants : 10, Similarité : 2.59%)
Région candidate trouvée à la position : 1097410 bits (Blocs correspondants : 10, Similarité : 2.59%)
Région candidate trouvée à la position : 1101840 bits (Blocs correspondants : 10, Similarité : 2.59%)
...
Région candidate trouvée à la position : 1029329620 bits (Blocs correspondants : 10, Similarité : 2.59%)
Temps d'exécution : 16.362129 sec
Estimation empirique de la complexité algorithmique, sur le temps d’exécution
import matplotlib.pyplot as plt
def mesurer_temps(n_bits, petit_length=100, taille_bloc=5, min_correspondances=10, max_ecart=6):
grand = np.random.choice(['0', '1'], size=n_bits)
petit = np.random.choice(['0', '1'], size=petit_length)
start = time.time()
regions_correspondantes(grand, petit, taille_bloc=taille_bloc,
min_corr=min_correspondances, max_ecart=max_ecart)
return (time.time() - start) * 1000 # s --> ms
# ------------ Générer une série de tailles d'entrée multiples de 5 allant de 1000 à 500000, 30 échantillons.
tailles_entree = np.linspace(1000, 500000, 30, dtype=int)
tailles_entree = tailles_entree - (tailles_entree % 5)
# -------------Mesurer le temps pour chaque taille d'entrée
temps_mesures_ms = np.array([mesurer_temps(n) for n in tailles_entree])
for taille, t in zip(tailles_entree, temps_mesures_ms):
print(f"Input size: {taille} bits, Time: {t:.6f} ms")
# ----- Régression log-log pour estimer l'exposant asymptotique k
log_n = np.log(tailles_entree)
log_t = np.log(temps_mesures_ms)
#----- Ajustement d'une droite aux données (log_n, log_t)
coefficients = np.polyfit(log_n, log_t, 1)
pente = coefficients[0]
intercept = coefficients[1]
print(f"Exposant estimé (k) : {pente:.2f}")
# ----- Calcul des temps ajustés à partir de la droite de régression
log_t_ajuste = intercept + pente * log_n
temps_ajustes = np.exp(log_t_ajuste)
# -- Tracé des données en échelle log-log ainsi que la droite ajustée
plt.figure(figsize=(10, 6))
plt.loglog(tailles_entree, temps_mesures_ms, 'bo', label='Données empiriques')
plt.loglog(tailles_entree, temps_ajustes, 'r-', label=f'Droite ajustée (k ≈ {pente:.2f})')
plt.xlabel("Taille d'entrée (bits)")
plt.ylabel("Temps d'exécution (ms)")
plt.title("Régression Log-Log pour estimer la complexité")
plt.legend()
plt.grid(True, which="both", ls="--")
plt.show()Input size: 1000 bits, Time: 0.602722 ms
Input size: 18205 bits, Time: 0.568390 ms
Input size: 35410 bits, Time: 0.933409 ms
Input size: 52620 bits, Time: 6.243229 ms
Input size: 69825 bits, Time: 2.574682 ms
Input size: 87030 bits, Time: 2.814054 ms
Input size: 104240 bits, Time: 2.959967 ms
Input size: 121445 bits, Time: 3.219604 ms
Input size: 138655 bits, Time: 3.438234 ms
Input size: 155860 bits, Time: 2.760172 ms
Input size: 173065 bits, Time: 2.748489 ms
Input size: 190275 bits, Time: 4.421711 ms
Input size: 207480 bits, Time: 5.502701 ms
Input size: 224685 bits, Time: 8.026600 ms
Input size: 241895 bits, Time: 6.815910 ms
Input size: 259100 bits, Time: 4.650593 ms
Input size: 276310 bits, Time: 3.922224 ms
Input size: 293515 bits, Time: 5.002975 ms
Input size: 310720 bits, Time: 5.844593 ms
Input size: 327930 bits, Time: 6.677866 ms
Input size: 345135 bits, Time: 7.516384 ms
Input size: 362340 bits, Time: 6.935120 ms
Input size: 379550 bits, Time: 6.588459 ms
Input size: 396755 bits, Time: 6.804943 ms
Input size: 413965 bits, Time: 7.915497 ms
Input size: 431170 bits, Time: 6.919146 ms
Input size: 448375 bits, Time: 9.229660 ms
Input size: 465585 bits, Time: 6.771803 ms
Input size: 482790 bits, Time: 7.102966 ms
Input size: 500000 bits, Time: 9.367228 ms
Exposant estimé (k) : 0.50
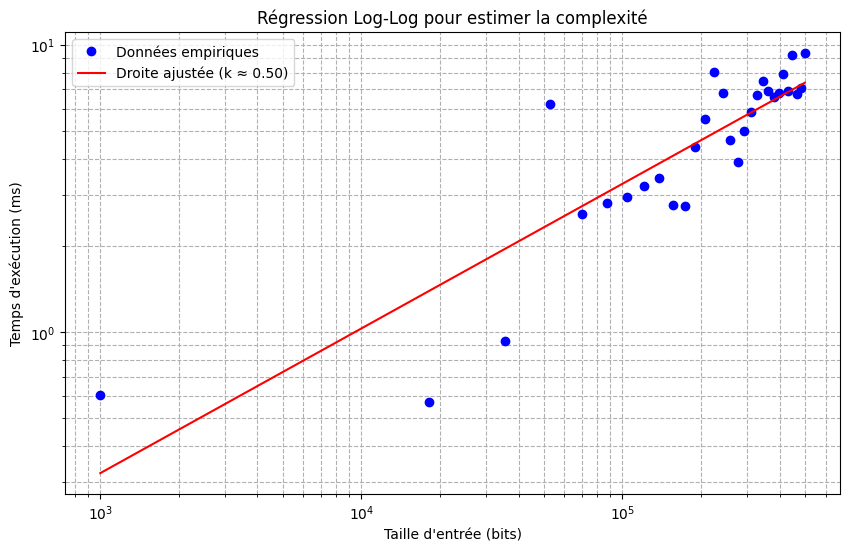
Donc l’algorithme avec la manière dont il est compilé semble avoir une complexité en temps meilleure que O(n) et au mieux